
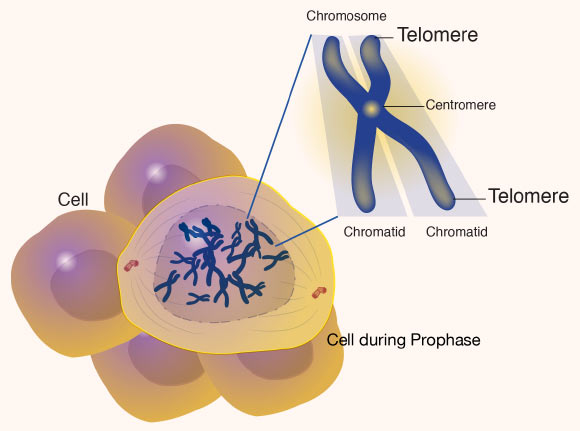
A team of U.S. and U.K. scientists has generated the end-to-end gapless DNA sequence of the human X chromosome.
Source: Sci News
After nearly two decades of improvements, the reference sequence of the human genome is the most accurate and complete vertebrate genome sequence ever produced.
However, there are hundreds of gaps or missing DNA sequences that are unknown.
These gaps most often contain repetitive DNA segments that are exceptionally difficult to sequence. Yet, these repetitive segments include genes and other functional elements that may be relevant to human health and disease.
Because a human genome is incredibly long, consisting of about 6 billion bases, DNA sequencing machines cannot read all the bases at once.
Instead, geneticists chop the genome into smaller pieces, then analyze each piece to yield sequences of a few hundred bases at a time. Those shorter DNA sequences must then be put back together.
“Our project was made possible by new sequencing technologies that enable ‘ultra-long reads,’ such as the nanopore sequencing technology,” said lead author Dr. Karen Miga, a research scientist at the UC Santa Cruz Genomics Institute.
“Repeat-rich sequences were once deemed intractable, but now we’ve made leaps and bounds in sequencing technology,” she added.
“With nanopore sequencing, we get ultra-long reads of hundreds of thousands of base pairs that can span an entire repeat region, so that bypasses some of the challenges.”
“This accomplishment begins a new era in genomics research,” said Dr. Eric Green, director of the National Human Genome Research Institute (NHGRI), part of the National Institutes of Health (NIH).
“The ability to generate truly complete sequences of chromosomes and genomes is a technical feat that will help us gain a comprehensive understanding of genome function and inform the use of genomic information in medical care.”
Of the 24 human chromosomes (including X and Y), the researchers chose to complete the X chromosome sequence first, due to its link with a myriad of diseases, including hemophilia, chronic granulomatous disease and Duchenne muscular dystrophy.
Humans have two sets of chromosomes, one set from each parent. For example, biologically female humans inherit two X chromosomes, one from their mother and one from their father. However, those two X chromosomes are not identical and will contain many differences in their DNA sequences.
In the study, the authors did not sequence the X chromosome from a normal human cell. Instead, they used a special cell type – one that has two identical X chromosomes.
Such a cell provides more DNA for sequencing than a male cell, which has only a single copy of an X chromosome. It also avoids sequence differences encountered when analyzing two X chromosomes of a typical female cell.
To finish the X chromosome, the team had to close all 29 remaining gaps in the current reference.
Two segmental duplications were resolved with ultra-long nanopore reads that completely spanned the repeats and were uniquely anchored on either side.
The remaining break was at the centromere, a notoriously difficult region of repetitive DNA found in every chromosome.
In the X chromosome, the centromere encompasses a region of highly repetitive DNA spanning 3.1 million base pairs.
Dr. Miga and colleagues were able to identify variants within the repeat sequence to serve as markers, which they used to align the long reads and connect them together to span the entire centromere.
“For me, the idea that we can put together a 3-megabase-size tandem repeat is just mind-blowing,” Dr. Miga said.
“We can now reach these repeat regions covering millions of bases that were previously thought intractable.”
The next step was a polishing strategy using data from multiple sequencing technologies to ensure the accuracy of every base in the sequence.
“We used an iterative process over three different sequencing platforms to polish the sequence and reach a high level of accuracy,” Dr. Miga said.
“The unique markers provide an anchoring system for the ultra-long reads, and once you anchor the reads, you can use multiple data sets to call each base.”
Nanopore sequencing, in addition to providing ultra-long reads, can also detect bases that have been modified by methylation, an epigenetic change that does not alter the sequence but has important effects on DNA structure and gene expression.
By mapping patterns of methylation on the X chromosome, the scientists were able to confirm previous observations and reveal some intriguing trends in methylation patterns within the centromere.
The results were published online July 14, 2020 in the journal Nature.
Source: Sci News


































Leave a Comment
You must be logged in to post a comment.